疾患遺伝子:病気は遺伝によって決まるのか
ヒトゲノム human genome

遺伝子の転写 transcription
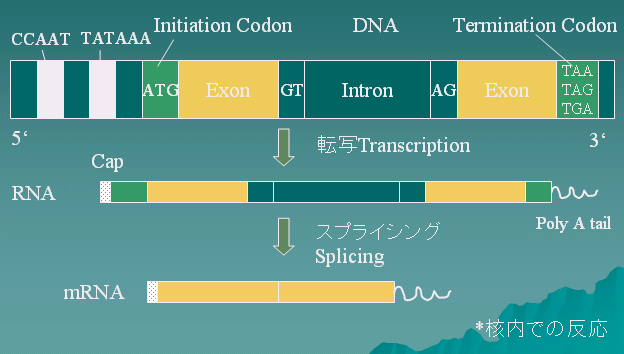
遺伝子の翻訳 translation
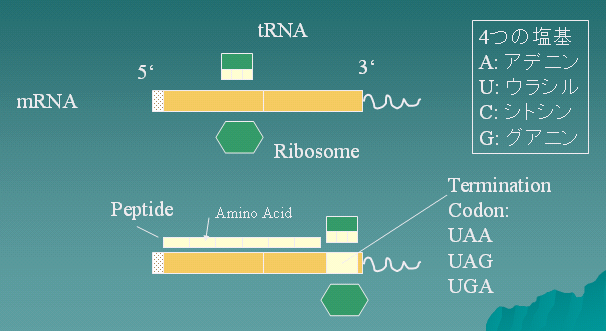
遺伝子コード genetic code

ハイブリダイゼーション hybridization

サザンブロット法

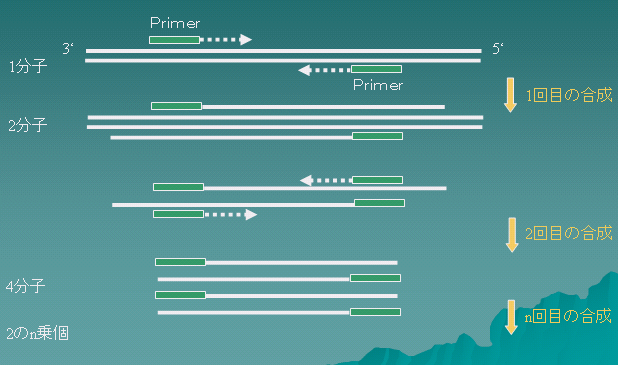
Polymerase Chain Reaction (PCR) ポリメラーゼ連鎖反応
極微量の(1 mlあたり100分子以下)DNA断片を増幅して検出することが出来る。
プライマー Primer DNAを用いて特定の遺伝子配列の部分を増幅することが出来る。
抽出したDNAサンプルにプライマーを混合してハイブリダイズさせ、DNAポリメラーゼ(合成酵素)を反応させる→温度を上昇させて反応を止めてDNAを乖離させる→これを繰り返す:n回で2nに増える
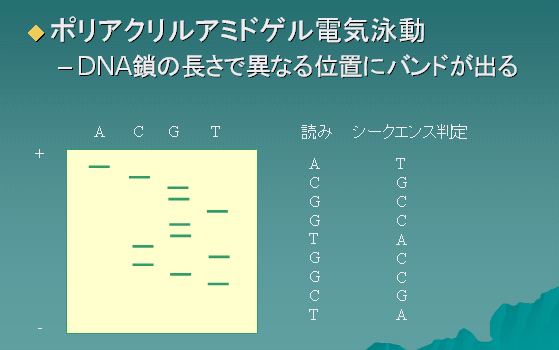
DNA塩基配列決定法
Dideoxyシークエンス法

ポリアクリルアミドゲル電気泳動による塩基配列の読み
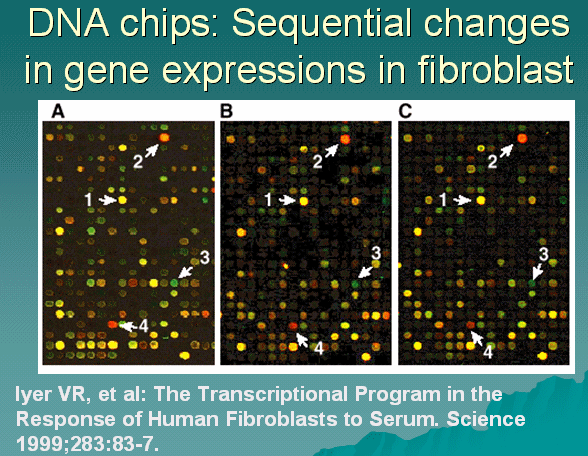
DNAマイクロアレイ(DNAチップ、ジーンチップ)
ヒト疾患遺伝子
Menderian Inheritance in Man(ヒト遺伝子病のカタログ)
13,761のメンデル形質が登録されている(2002.7.5現在)
遺伝子と臨床症状は1対1で対応しない
一般的な疾患
環境要因
複数の遺伝子の組み合わせが複雑に関与
Oligogenic Disorder
Polygenic Disorder
臨床診断と遺伝子診断
臨床症状(自覚症状と他覚症状)の出現頻度が疾患で異なる → 症状の組み合わせパターンで診断する。
疾患に特有の(特異的な)遺伝子の配列が存在する ?→ 遺伝子の配列によって診断する。
いずれも100%の感度・特異度ではない突然変異の分類
・欠失 Deletion
・挿入 Insertion
・1塩基置換
-ミスセンス突然変異:1個のAAが別のAAに置換
-ナンセンス突然変異:1個のAAが終止コドンになる
-スプライス部位突然変異:スプライシングのシグナルを出現あるいは消失させる
・フレームシフト:欠失、挿入,スプライシングエラーにより生ずる機能獲得性突然変異と疾患

機能喪失性突然変異と疾患

遺伝子量効果と疾患

同じ遺伝子の異常が複数の疾患で認められる

染色体数の異常
倍数体
3倍体(69、XXX,XXY,XYY):全妊娠例の3%、ほとんど死産、生存率ゼロ
異数体
-常染色体
・ナリソミー:相同染色体の1対が欠損、着床前に死亡
・モノソミー:1本の染色体が欠如、胚の時期に死亡
・トリソミー:1本の過剰染色体がある、通常胚または胎児期に死亡:トリソミー21=ダウン症候群、トリソミー18=エドワード症候群、トリソミー13=Patau症候群
-性染色体
・XXX,XXY,XYY:比較的問題少ない
・45、X:99%は自然流産、生存者は不妊、知能は平均的SNP
SNP (single nucleotide polymorphism)スニップ、 1塩基遺伝子多型性
ヒトゲノム30億塩基対の内、99.9%は人類共通
残り、0.1%、すなわち約300万塩基対は個人個人によって異なる
同じ遺伝子の同じ部分を個人個人で比べた場合、1塩基だけが異なっていることがある
人口の1%以上で異なっている場合、スニップという
スニップが疾患感受性と関係している可能性がある
疾患関連遺伝子同定の問題点
ヒトゲノムの遺伝情報と一般的疾患の関連についての研究は急速に発展している。近い将来、一般臨床の場で、診断、治療の選択、発症予防などに際して、ヒトゲノム配列情報が用いられるようになるであろう。疾患とゲノム配列の関連を証明する方法として、2つの研究法が用いられている。ひとつは、連鎖解析 Linkage analysisであり、もうひとつが関連解析 Association studyである。後者について簡単に解説する[1]。
関連解析では症例対照研究と家系解析の2つの方法が用いられている。症例対照研究は通常の症例対照研究と全く同じ研究手法である。症例にマッチングさせた対照を集め、両群でのさまざまな配列の頻度を比較する。この際に、対照群の人種構成が異なっていたような場合、人種差を規定している遺伝子の頻度に差があるのは当然であるが、それを疾患と関連していると取り違える可能性がある。そこで、適切な層別化が行なわれることがある[2]。家系解析では、症例の属する家系で対照を選択することによって、疾患関連配列以外の差を小さく出来る。
関連解析では、あらかじめ疾患と関連していると想定される候補遺伝子を見出す必要がある。すべての遺伝子の生物学的役割や機能が分かっているわけではないので、症例群と対照群で頻度の差が見つかっても、それが偶然による可能性が否定できないことが多い。すなわち、単に頻度だけを比較して、差を見出しても、生物学的には無関係の配列を見ているかもしれない。人類のゲノムの塩基配列はほぼ共通であり、ランダムに抽出された2人のゲノムを比較した場合、塩基が異なる点は、1250塩基に1つしかない。タンパクをコードする遺伝子だけを見ると、さらに少なく、2000塩基に1つである[3]。さらに、2人のゲノム間で異なる配列の大部分は、ヒト集団の中で、共通に認められるものであり、ゲノムの塩基配列の相違は一般人口の1%以上で認められるものである[4]。したがって、患者1人とその疾患の無い者1人のゲノム配列を比較して、異なる点を拾い上げても、ほとんどは疾患と関係が無い差である。
ヒトゲノムには、10,000,000個の一塩基多型 (Single-nucleotide polymorphisms、SNPスニップ)があると推定されているが、この中で、例えば、20が実際に疾患と関連していると仮定しよう。症例対照研究で、P値0.05で有意であると判定した場合、1千万個のSNPの内、その20分の1、すなわち、500,000が偽陽性、その内たった20個が真の陽性であるという結果を得ることになる。したがって、出現頻度に有意差が認められた50万個のほとんどが、偶然P値が小さくなる結果が得られただけであって、疾患とは関係が無いということになる。もし、P値が10-6以下の場合に有意であると判定した場合には、偽陽性は10個だけになるが、真の20個のSNPを検出するには、極めて大きなサンプルサイズが必要になる[5]。
このように、比較する変数が極めて多いため、関連解析では、統計学的なエラーを起こす可能性が高い。したがって、ひとつの研究で示された関連が、検証研究で裏付けられる確率が低くなり、実際にそれが証明されている。4つ以上の研究が行なわれた166件の関連の中で、結果がどの研究でも同じだったものは、6件しかなかったことが報告されている[6]。このような結果になるのは、それぞれの研究が、サンプルサイズが不十分であったり、偽陽性を除外するための基準が甘いことが原因である[7]。The false positive report probability (FPRP)による方法が最近紹介されている[8]。
文献
[1] Rosand J, Altshuler D: Human genome sequence variation and the search for genes influencing stroke. Stroke 2003;34:2512-7.
[2] Ozaki K, Ohnishi Y, Iida A, Sekine A, Yamada R, Tsunoda T, Sato H, Hori M, Nakamura Y, Tanaka T: Functional SNPs in the lymphotoxin-alpha gene that are associated with susceptibility to myocardial infarction. Nat Genet 2002;32:650-4.
[3] Cargill M, Altshler D, Ireland J, Sklar P, Ardlie K, Patil N, Shaw N, Lane CR, Lim EP, Kalyanaraman N, et al: Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet 1999;22:231-8.
[4] Marth G, Yeh R, Minton M, Donaldson R, Li Q Duan S, Davenport R, Miller RD, Kwok PY: Single-nucleotide polymorphisms in the public domain: how useful are they? Nat Genet 2001;27:371-2.
[5] Dahlman I, Eaves IA, Kosy R, Morrison VA, Heward J, Gough SC
Allahabadia A, Franklyn JA, Tuomilehto J, Tuomilehto-Wolf E, et al: Parameters for relioable results in genetic association studies in common disease. Nat Genet 2003;33:457-8.
[6] Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K: A comprehensive review of genetic association studies. Genet Med 2002;4:45-61.
[7] Lohmueller KE, Pearce CL, Pkike M, Lander ES, Hirschhorn JN: Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet 2003;33:177-82.
[8] Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, Rothman N. J Natl Cancer Inst 2004;96:434-42.